






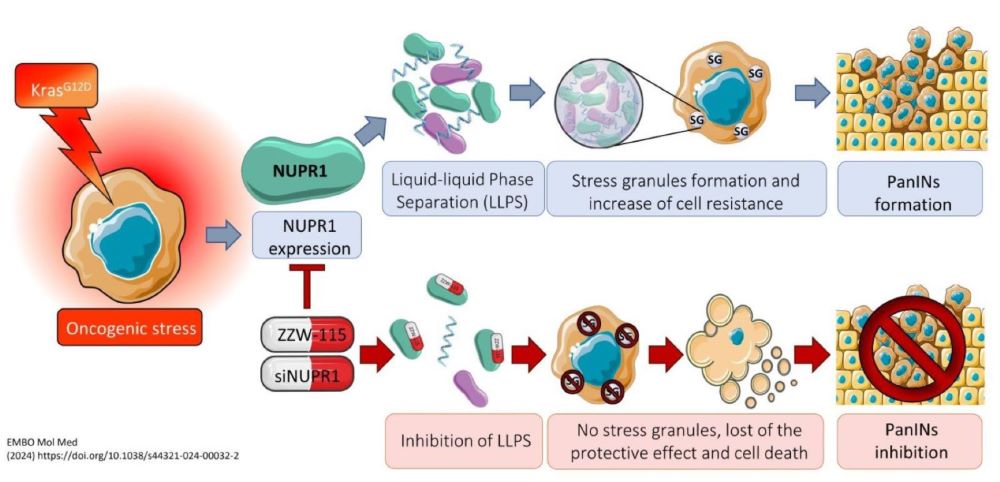













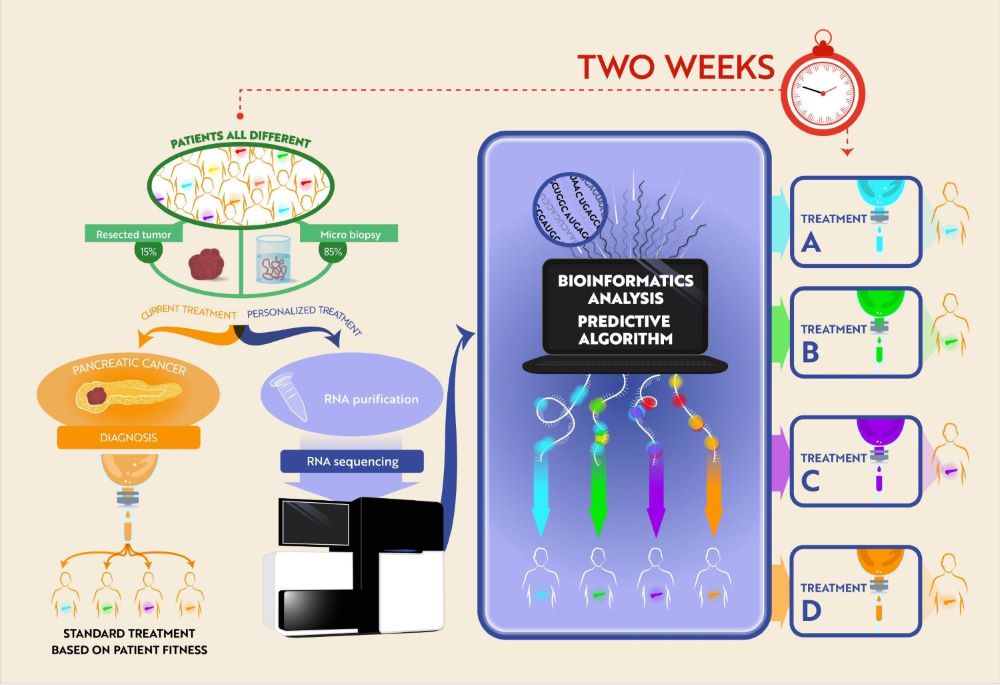
Mass Cytometry Reveals Classical Monocytes, NK Cells, and ICOS+ CD4+ T Cells Associated with Pembrolizumab Efficacy in Patients with Lung Cancer.
Rochigneux P, Lisberg A, Garcia A, Granjeaud S, Madroszyk A, Fattori S, Gonçalves A, Devillier R, Maby P, Salem N, Gorvel L, Chanez B, Gukasyan J, Carroll J, Goldman J, Chretien AS, Olive D, Garon EB